RESUMEN
Las enfermedades infecciosas constituyen una de las mayores amenazas a la salud humana y animal a nivel global, generando una enorme carga en cuanto a morbimortalidad. Para luchar contra este problema se hace necesario el desarrollo de técnicas de diagnóstico microbiológico que permitan establecer la etiología de la enfermedad de una manera sencilla y fiable. De acuerdo a los criterios ASSURED de la OMS1, la técnica de diagnóstico ideal debería ser barata, fiable, proporcionando un resultado rápido y que no requiriese de material o técnicas complejas. Se puede afirmar que hasta la fecha ninguna técnica disponible cumple con todas estas características2, lo que hace necesario que sigamos profundizando en el conocimiento actual.
Los sistemas bacterianos de defensa CRISPR/Cas fueron descubiertos hace más de tres décadas3, pero solo recientemente usando ingeniería genética han sido adaptados para utilización en humanos. El empleo de esta tecnología para la edición de genes hace posible su uso en la terapia de las enfermedades hereditarias, entre otras3. Más reciente aún ha sido el diseño de dicha tecnología para conseguir la detección de ácidos nucleicos; así en los últimos años hemos sido testigos de un verdadero auge en el desarrollo de técnicas diagnósticas tales como CONAN, SHERLOCK, DETECTR y otras muchas. Incluso, en referencia al campo de las enfermedades infecciosas, se ha aplicado esta tecnología para estudiar las interacciones huésped-receptor, habiendo demostrado ser de gran utilidad para el estudio de la causalidad de determinadas enfermedades.
La gran ventaja del uso de estas técnicas consiste en una gran sensibilidad y especificidad, equiparable en algunos casos a los resultados que podemos obtener con técnicas como la PCR, pero que además no requiere del uso de equipamiento sofisticado. Muchas de ellas han sido empleadas para la detección de determinados virus, como pueden ser los trabajos para conseguir la diferenciación entre HPV16 y HPV18, con excelentes resultados en cuanto a tiempo de resultado. Por supuesto, estas técnicas también presentan una serie de inconvenientes que se deben tener en cuenta a la hora de valorar un posible y futuro uso en el diagnóstico etiológico de enfermedades infecciosas.
El objetivo de este trabajo, una revisión bibliográfica, será recopilar y analizar el estado del conocimiento actual del desarrollo de técnicas diagnósticas microbiológicas basadas en la tecnología CRISPR-Cas, enfatizando sus ventajas e inconvenientes con respecto a otras técnicas, para poder ofrecer una panorámica general del posible futuro de estos diseños diagnósticos.
INTRODUCCIÓN
Las enfermedades infecciosas suponen una de las mayores cargas en cuanto a morbimortalidad en todo el mundo4. Para combatirlas, se han desarrollado múltiples técnicas de diagnóstico, siendo las técnicas de diagnóstico molecular como la Reacción en Cadena de la Polimerasa (PCR, por sus siglas en inglés) uno de los mayores avances en diagnóstico de las últimas décadas. Esta técnica ha sido ampliamente usada durante la pandemia reciente por el virus SARS-CoV25, poniendo a prueba la capacidad logística de muchos laboratorios de Microbiología Cínica en todo el planeta. Este método diagnóstico posee una serie de ventajas, pero también un conjunto de inconvenientes como pueden ser los requerimientos de un personal cualificado y de un equipamiento sofisticado y costoso6. Es por eso que se hacen esfuerzos en investigación para desarrollar nuevas técnicas de diagnóstico molecular, como las técnicas de amplificación isotérmica, que sirvan como alternativas a la PCR. Esta búsqueda de alternativas resulta especialmente importante en países o regiones en vías de desarrollo, en los cuales un requerimiento de reactivos y materiales que tengan un alto precio puede frenar o imposibilitar el diagnóstico de enfermedades infecciosas, dificultando así el control de brotes infecciosos6,7.
Hace aproximadamente 30 años un investigador alicantino descubrió un sistema bacteriano de defensa antiviral que proporciona una suerte de “inmunidad adaptativa”3 y que está presente en la mayor parte de las arqueas y en la mitad de las bacterias. El sistema fue denominado CRISPR-Cas y sentó las bases de uno de los mayores avances en Biología de los últimos tiempos: el uso de estos mismos sistemas para realizar procesos de ingeniería genética, pudiendo usarse en células de mamíferos, entre ellas, claro está, células humanas8,9,10. Estos sistemas son altamente precisos y ofrecen la posibilidad de modificar virtualmente cualquier secuencia de DNA o RNA en nuestro cuerpo. Sin embargo, la aplicación de esta tecnología no ha quedado circunscrita únicamente al campo de la ingeniería genética y el tratamiento de enfermedades genéticas, sino que se buscan desde hace años posibles aplicaciones de los mismos en el campo de la terapéutica de las enfermedades infecciosas y en el diagnóstico de las mismas11. Desde 2016, momento de su surgimiento, los métodos de detección de ácidos nucleicos basados en los sistemas CRISPR-Cas se han multiplicado cada vez con mayor celeridad. Estos métodos ofrecen en algunos casos unos valores de sensibilidad y especificidad similares a los de la PCR, con la ventaja añadida de una menor complejidad y un menor requerimiento de equipamiento. Esto ha llevado a algunos autores2,11,12 a proponerlos como test diagnósticos Point-of-Care (POC) que puedan introducirse en la práctica clínica habitual.
Este trabajo comienza con una serie de generalidades sobre la biológica básica de estos sistemas, con el fin de entender sus complejos procesos y poder comprender cómo pueden usarse para aplicaciones humanas.
BIOLOGÍA DE LOS SISTEMAS CRISPR-CAS
Desde hace millones de años se lleva desarrollando lo que algunos autores13 han dado en llamar una “carrera armamentística” entre las bacterias y los virus que las infectan, los bacteriófagos. Dicha carrera involucra diversos sistemas de defensa que las bacterias han desarrollado para poder defenderse de las infecciones producidas por dichos agentes, además de otros elementos genéticos móviles (MGEs, por sus siglas en inglés) como los transposones o los plásmidos13,14,15. Entre los mecanismos de defensa más extendidos se encuentran los sistemas enzimáticos de Restricción y Modificación (R-M)13. Estos sistemas reconocen y discriminan entre secuencias foráneas invasoras y secuencias bacterianas gracias a que estas últimas se encuentran modificadas, generalmente mediante metilación (aunque las bacterias pueden emplear otros tipos de modificaciones). Una vez discriminada la secuencia y reconocida como extraña, estas enzimas proceden a realizar un corte de las mismas, para evitar así la progresión de la infección. Existen otros sistemas como BREX16 o DISARM17 que funcionan de manera similar. Estos sistemas de detección de ácidos nucleicos constituirían una especie de “inmunidad innata” en las bacterias, dado que funcionan sin una memoria inmunológica.
Los sistemas procariotas CRISPR-Cas fueron descubiertos en 1989 y a principios de la década de los 90 se postuló la hipótesis de que eran sistemas de defensa bacterianos que proporcionaban “inmunidad innata” a la célula bacteriana3. En el año 2006 llegó la confirmación experimental de tal hipótesis; las bacterias eran capaces de generar una memoria de determinadas secuencias y trasmitirlas a su descendencia. Desde entonces se han ido ampliando los conocimientos acerca de estos sistemas de defensa bacterianos, pero el pistoletazo de salida a la ingeniería genética basada en CRISPR-Cas lo darían en 2012 las científicas Emmanuelle Charpentier y Jennifer Doudna3,9, quienes más tarde recibieron el premio Nobel en 2020. Estas científicas describieron el sistema CRISPR-Cas9, el primero en ser utilizado para tales fines, como una herramienta de edición de gran precisión, mucho más eficaz que otros sistemas que se venían usando hasta la fecha, como las nucleasas con dedos de zinc (ZFNs por sus siglas en inglés) o las nucleasas tipo activadores de transcripción (TALENs por sus siglas en inglés)9. Quedaba así abierta la puerta a una verdadera revolución, basada toda ella en un principio muy simple, aunque con gran complejidad intrínseca: una secuencia de RNA complementaria a una secuencia objetivo guiaría a una nucleasa, Cas9, para hacer cortes en dicha diana. Una vez producido el corte, se usarían los mecanismos propios de la célula receptora para la reparación de las roturas de doble hebra.
CRISPR (Clustered Regularly Interspaced Palindromic Repeats, que se puede traducir al español como repeticiones palindrómicas cortas regularmente espaciadas y agrupadas) se define, pues, como un sistema de inmunidad procariota basado en la adquisición de espaciadores para la defensa frente a material genético extracelular15. Su estructura consiste en repeticiones palindrómicas de 23 a 55 pares de bases, que se encuentran separadas por espaciadores, de 21 a 72 pares de bases. El locus CRISPR comprende la región cromosómica correspondiente al CRISPR array, la secuencia guía o leader y los genes Cas18. Lo que denominamos CRISPR array sería esa región genética correspondiente a los complejos repeticiones-espaciadores. Fueron precisamente estos espaciadores y su similitud con secuencias genéticas de bacteriófagos, plásmidos y transposones, la clave para comprender que este sistema aportaba un complejo mecanismo de inmunidad a la bacteria3. Los genes Cas (CRISPR associated genes), por otra parte, son genes situados en la vecindad de las secuencias CRISPR15. Su transcripción da origen a numerosas proteínas, también denominadas Cas, que poseen numerosas e importantes funciones en el proceso inmunológico del sistema.
Es lógico que todos estos sistemas presenten una gran diversidad18, dada la enorme variedad de bacteriófagos que pueden infectar a los procariotas. Podemos dividir los sistemas CRISPR-Cas en 2 clases basándonos en los genes Cas específicos de cada subtipo CRISPR-Cas, la similitud de secuencia entre las proteínas Cas y la organización de los genes en los loci CRISPR-Cas15,19. Los sistemas pertenecientes a la clase I, que a su vez se subdivide en 3 tipos (tipo I, tipo II y tipo III, que a su vez se subdividen en subtipos), se caracterizan por usar complejos efectores conformados por varias proteínas Cas. Son los más abundantes del total de loci CRISPR-Cas identificados en arqueas y bacterias, pero también son más complejos15,19. Por otra parte los sistemas de clase II (tipos II, V y VI) se caracterizan porque sus mecanismos de interferencia se componen de una sola proteína efectora. El esquema general de funcionamiento de estos sistemas, si bien pueden observarse múltiples modificaciones en la naturaleza, puede dividirse en 3 etapas: Adaptación, Síntesis del RNA CRISPR (crRNA) e Interferencia15.
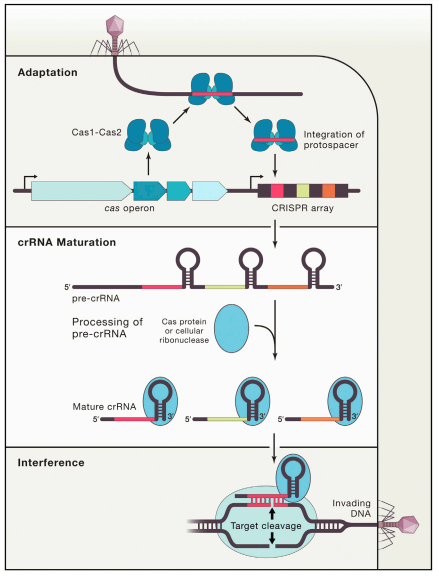
INTERFERENCIA
La interferencia sería el proceso mediante el cual se produce la destrucción de MGEs que invaden la bacteria15,19. Se trataría del paso final de este sistema de defensa y el que más interesa desde el punto de vista de la aplicación de la tecnología basada en CRISPR-Cas. Tendría lugar cuando un bacteriófago u otro MGE que ya hubiese infectado a la bacteria, volviese a infectar al microorganismo.
Una propiedad fundamental del sistema CRISPR-Cas es su capacidad de discernir entre moléculas de ácidos nucleicos invasoras y secuencias genómicas propias, con el fin de evitar la autoinmunidad15,19. Casi todos los sistemas CRISPR-Cas caracterizados (exceptuando el tipo III) poseen un mecanismo que involucra el reconocimiento, por parte de la maquinaria de interferencian y la maquinaria de adaptación, de una secuencia corta de nucleótidos, denominada motivo adyacente al protoespaciador (PAM, por las siglas en inglés de Protospacer Adjacent Motif). La presencia de una secuencia PAM en la secuencia invasora y su ausencia en secuencias genómicas bacterianas consigue evitar la autoinmunidad. Sin mecanismos discriminadores como este y otros, la maquinaria de interferencia podría acabar destruyendo el propio CRISPR array bacteriano15.
LA INTERFERENCIA EN LOS SISTEMAS CRISPR-CAS DE CLASE 1
Los sistemas CRISPR-Cas de tipo I, pertenecientes a la clase 1, son los más diseminados entre las especies bacterianas15,19, siendo los más estudiados, especialmente los identificados en ciertas cepas de Escherichia coli. La maquinaria de interferencia en este caso se compone de un complejo enzimático denominado Cascade20 (siglas de CRISPR-associated complex for antiviral defense), que contiene múltiples proteínas Ca (de entre las que destacan Cas5, Cas6, Cas7, Cas8 y Cas11), y que están unidas a una molécula de RNA. Esta molécula de RNA se conoce como crRNA (RNA CRISPR) y procede de los loci de CRISPR, de modo que alberga secuencias de espaciadores concretas. Esta secuencia actuaría como RNA guía para el complejo de ataque, ya que guarda homología con secuencias concretas de MEGs que ya hayan invadido anteriormente a la bacteria y que esta ha memorizado en forma de espaciador, alojándolas en un CRISPR array.
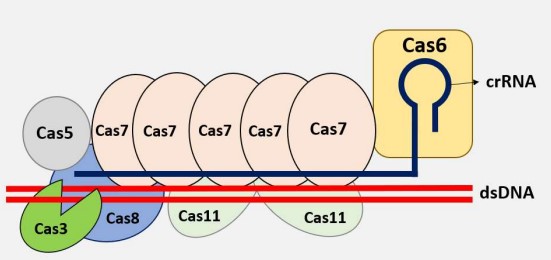
Mediante algunas de las proteínas del complejo Cascade se procederá a buscar el reconocimiento de PAM en una secuencia invasora si se ha producido una infección20; si dicha estructura está presente se produce el desenrollamiento inicial de DNA y la unión del crRNA a su secuencia de DNA complementaria invasora, en caso de estar ésta también presente y existir homología. Se forma así una estructura R-loop y se recluta a otra proteína, la nucleasa Cas320,21. Esta proteína es una pieza fundamental de este proceso15, ya que es la que lleva a cabo la rotura de la cadena de DNA complementaria del genoma viral, la que no está unido al complejo Cascade, provocando de esta manera la destrucción del genoma invasor y frenando la progresión de la infección.
En caso de los sistemas CRISPR-CAS de tipo III, también pertenecientes a la clase 1, emplean un complejo similar a Cascade15,19, puesto que también se caracteriza por presentar múltiples proteínas en su complejo de interferencia. En el caso del subtipo III-A dicho complejo se denomina Csm y en el caso de III-B, se denomina Cmr19. Aunque presentan una gran similitud en cuanto a estructura y funcionamiento con los sistemas tipo I, los sistemas CRISPR-Cas de este tipo tienen una serie de particularidades que merece la pena destacar: tienen como substratos tanto cadenas de RNA como cadenas de DNA22 (dependiendo en este último caso de la transcripción de la secuencia objetivo), además de que no requieren el reconocimiento de PAM15,22 para poder ejercer su función, constituyendo esto una especie de excepción a la regla. Otra característica esencial de los sistemas tipo III es que sería en este caso la proteína Cas10, perteneciente al complejo Csm, la que realiza el corte de las cadenas de DNA invasor y también es esta la proteína que lleva a cabo una conversión de ATP a cOA (siglas en inglés de cyclic oligo-(A)-nucleotides)22, que actúa de segundo mensajero para activar la RNasa no específica Csm6. Dicha enzima, si bien no forma parte del complejo Csm, funciona como una ayuda auxiliar en la degradación de material genético externo, ya que se encargaría de la degradación no específica (es decir, no mediada por crRNA, sino que es una función intrínseca) de secuencias adyacentes. La producción de los segundos mensajeros por Cas10 depende de la unión del complejo Csm al RNA objetivo, de modo que cuanto más RNA objetivo esté presente, mayor degradación por Cas10 y mayor activación de Csm6, estableciéndose así un mecanismo de regulación. Es importante destacar esta cualidad del sistema CRISPR-Cas tipo III, y más concretamente, destacar las propiedades de Csm6, dadas las implicaciones que estas han tenido en diseños recientes de técnicas diagnósticas basadas en la tecnología CRISPR-Cas.
Por último, los sistemas CRISPR-Cas de tipo IV no están muy estudiados, son precarios y varios puntos de su funcionamiento permanecen desconocidos15,19.
LA INTERFERENCIA EN LOS SISTEMAS DE CLASE 2
Como se mencionó anteriormente, los sistemas pertenecientes a esta clase presentan una única proteína efectora, grande y con varios dominios, unida a un crRNA como maquinaria de interferencia15,19. Esta característica hace que estos sistemas sean menos complejos, lo que los ha convertido en los sistemas más estudiados para ser empleados en la tecnología de ingeniería genética y para otras aplicaciones. Sus loci también son más uniformes que los de la Clase 123.
En el caso de los sistemas CRISPR-Cas tipo II, Cas9 es la nucleasa tipo RuvC que forma parte de la maquinaria de interferencia23,24. Además, Cas9 posee otras funciones importantes como la maduración del crRNA15. Una estructura importante de estos sistemas y cuyo descubrimiento supuso un avance fundamental en el estudio de estos sistemas, es el trans-activating crRNA (tracrRNA)15,19,23,24, una pequeña molécula de RNA que posee complementariedad a regiones del crRNA y que es necesaria para formar la maquinaria de interferencia. Así pues, la proteína Cas9 se uniría a un RNA dual maduro conformado por la unión de tracrRNA y crRNA (tracrRNA:crRNA). Cas9 lleva a cabo el reconocimiento de PAM, permitiendo la unión de tracrRNA:crRNA a la secuencia concreta de DNA, lo que originaría un corte en ambas cadenas de DNA por parte de Cas9.
El tipo II es, con diferencia, el sistema CRISPR-Cas que mayor interés ha generado desde el principio en la investigación de ingeniería genética y otros usos8,9,10. Cas9 es una de las proteínas más estudiadas de este sistema, habiendo sido caracterizada en su totalidad, conociéndose sus lóbulos o regiones de actuación con gran fiabilidad, un avance en investigación asombroso y que se ha producido en muy escaso margen de tiempo.
Es importante comentar que la tecnología basada en CRISPR-Cas9 emplea una molécula RNA quimérica denominada single-guide RNA o sgRNA9,10, que combina crRNA con tracrRNA. Esto simplifica mucho el uso de esta maquinaria en diversas aplicaciones de ingeniería, ya que reducimos el número de componentes que usemos a solo 2: Cas9 y sgRNA. La clave estaría en crear una secuencia sgRNA deseada que guiase a Cas9 hacia una secuencia diana que se pretendiese modificar.
Por otra parte, debemos tener presente que existen múltiples ortólogos de Cas923, así como de otras proteínas Cas, dada la gran variabilidad que pueden presentar estos sistemas entre especies bacterianas. La función será la misma en los diferentes ortólogos, pero diferirán en tamaño molecular y en otras características que puede hacer que unas u otras sean más interesantes en función de la aplicación que llevemos a cabo. La proteína Cas9, obtenida de Streptococcus pyogenes (abreviada como SpCas9) es una de las más usadas9,10.
Los sistemas CRISPR-Cas tipo V se subdividen en V-A, V-B y V-C, que a su vez se caracterizan por sus proteínas efectoras: Cas12a, Cas12b y Cas12c, respectivamente15,19. Estas enzimas no requieren tracrRNA para su cumplir con su función, estando conformada la maquinaria de interferencia únicamente por Cas12 unido a crRNA, que se dirige hacia su objetivo: DNA de doble cadena25. Si Cas12 es capaz de reconocer PAM, se podría producir la unión de crRNA con su DNA diana si hay homología y Cas12 procedería entonces a realizar cortes en las dos hebras de DNA de manera escalonada25,26. Recientemente se han descubierto las proteínas Cas1427, también pertenecientes a este tipo V, que son capaces de cortar cadenas simples de DNA sin el reconocimiento previo de PAM. Importante destacar que algunas proteínas Cas12 y Cas14 presentan una peculiar característica llamada actividad colateral28; una vez que el complejo de interferencia queda unido a su DNA diana, estas proteínas no solo son capaces de cortar su secuencia diana, sino que pueden destruir cualquier molécula de DNA adyacente al sitio de reacción de una manera no específica, sin necesidad de crRNA.
Por último, los sistemas CRISPR-Cas tipo VI se caracterizan por las proteínas efectoras Cas13 en sus maquinarias de interferencia, unidas a una molécula de crRNA. Cas13 presenta como característica especial que sus dianas son cadenas simples de RNA, no DNA29. También es muy interesante resaltar que estas enzimas presentan una actividad RNasa colateral una vez que se han unido el crRNA a su secuencia diana2,11,15,. De este modo se producirían cortes en tal secuencia y se degradaría de manera indiscriminada cualquier secuencia de RNA adyacente, incluso las del propio hospedador. Esta particularidad de Cas13 podría indicar que estas enzimas actúan como el detonante del suicidio del microorganismo (o su entrada en letargo) para evitar que el bacteriófago pueda acabar su ciclo lítico e infectar a otras células de la población bacteriana29. Como en el tipo V, no requiere tracrRNA, además de no requerir PAM, constituyendo otra excepción a la regla. Este tipo de sistema CRISPR-Cas deben reconocer una secuencia denominada PFS (protoespacer flanking site) para ejercer su acción, a la manera de PAM, pero este fenómeno se ha descubierto solo en algunos ortólogos y no parece que esté muy extendido15.

Por supuesto, todos estos sistemas de interferencia aquí señalados presentan gran número de variaciones19, siendo la diversidad mucho mayor de lo expresado aquí.
BIOGÉNESIS DEL crRNA
Como se ha comentado en el apartado anterior, la maquinaria de interferencia se compone no solo de proteínas Cas, sino que se precisa de un RNA que actúe de guía y que contenga secuencias que guarden una homología con el material genético de MEGs invasores15,19. Esta etapa de biogénesis de crRNA es la etapa de expresión, cuando el CRISPR array se transcribe dando lugar a una larga secuencia de RNA (conocida como pre-crRNA)15. Este pre-crRNA debe sufrir un proceso de maduración, que tiene como objetivo liberar secuencias crRNA constituidas por la secuencia de un solo espaciador y fragmentos de las repeticiones adyacentes, a las cuales se van a unir proteínas Cas para formar los complejos efectores descritos en el apartado anterior.
En los sistemas CRISPR-Cas de clase 1 el proceso de maduración involucra una proteína denominada Cas630, que reconoce estructuras en horquilla del pre-crRNA (formadas gracias a las secuencias repetitivas palindrómicas del CRISPR) y se une a ellas. Una vez unida Cas6, procesa el pre-crRNA gracias que esta enzima posee actividad nucleasa, liberándose así crRNA maduro. Interesante destacar que Cas6 se queda unido a este RNA para servir como una especie de andamio en la formación del complejo Cascade15,30.
En cuanto a la biogénesis en los sistemas CRISPR-Cas de clase 2, los de tipo II también requieren tracrRNA para este paso31. Una vez que se ha unido dicho tracrRNA al crRNA, Cas9 estabiliza el dúplex y corta, con ayuda de la RNasa III, el precursor, liberando así crRNA maduro. En los tipos V y VI, tanto las proteínas Cas12 como las Cas13 poseen actividad nucleasa dual para el paso de interferencia y para poder procesar el pre-crRNA, de modo que son también estas enzimas las responsables de este proceso15.
ADAPTACIÓN
Es un paso fundamental, dado que es el momento de todo el proceso en el que se genera la denominada memoria inmunológica de la bacteria. Sucede cuando un MEG ataca a un procariota por primera vez, por lo que el microorganismo podría identificar un pre-espaciador en la secuencia del invasor, susceptible de convertirse en un espaciador que la bacteria incorporase entre las secuencias de repeticiones palindrómicas en su CRISPR array, con el fin de memorizarla para futuras infecciones15. El proceso por el cual un procariota decide que secuencias invasoras determinadas debe seleccionar para incorporarlas en CRISPR es complejo y se trata de un asunto que está sujeto a debate.
Las proteínas Cas1 y Cas2 son las piezas fundamentales de la maquinaria de adaptación, estando presente en la gran mayoría de los tipos de CRISPR-Cas32, aunque se presentan variedades y excepciones, y no entraña un gran interés el desarrollarlas aquí. Baste decir que estas proteínas forman un complejo que transfiere la secuencia a un extremo del CRISPR array, en una manera que es similar a la integración retroviral15,19,32. De este modo se podrían leer los sucesivos espaciadores como un historial de las infecciones pasadas sufridas por el microorganismo procariota.
LA DETECCIÓN DE ÁCIDOS NUCLEICOS MEDIANTE CRISPR-CAS
Las enfermedades infecciosas son un problema de salud pública que genera una gran morbimortalidad en todo el mundo. Como se señaló al comienzo de este escrito, es preciso el desarrollo de nuevos métodos diagnósticos que puedan solventar ciertos problemas asociados a las técnicas de las que disponemos hoy día. Es por eso que desde hace ya unos años, más concretamente desde 201633, numerosos grupos de investigación han centrado sus esfuerzos en convertir esta tecnología CRISPR-Cas en métodos de detección de ácidos nucleicos fiables.
El esquema básico de estos diseños diagnósticos es el empleo de un sgRNA que dirige a una proteína Cas (obtenida de alguna bacteria o arquea), o bien a una modificación o variante artificial de algunas de estas proteínas, hacia una secuencia diana. Una vez unido el sgRNA a dicha secuencia, se pondrán en marcha diversos mecanismos que permiten, mediante un sistema de lectura, poder determinar si la secuencia diana está presente o no2,11,12,34,35.
Los primeros métodos diagnósticos basados en CRISPR-Cas creados usaban el sistema tipo II para la detección de ácidos nucleicos11. Aunque existen diversos diseños, uno de los primeros usa una proteína Cas9 inactiva (dCas9, por dead Cas9), la cual reconoce PAM en la secuencia diana, permitiendo la unión de sgRNA pero no se produce ningún corte en la secuencia al haber sido inactivada33. Así pues, en este diseño dos dCas9 se fusionan artificialmente cada una con un dominio de enzima luciferasa, y los sgRNA dirigen el complejo hacia dos regiones adyacentes la una de la otra dentro de una secuencia diana. Si dicha secuencia está presente, se producirá por proximidad la reconstitución de la enzima luciferasa, restaurando su actividad. Si hay luciferina en el medio, la actividad de esta enzima generará una seña de fluorescencia que puede ser medida. Este método se empleó para la detección de DNA de Mycobacterium tuberculosis, con buenos resultados de sensibilidad y especificidad.

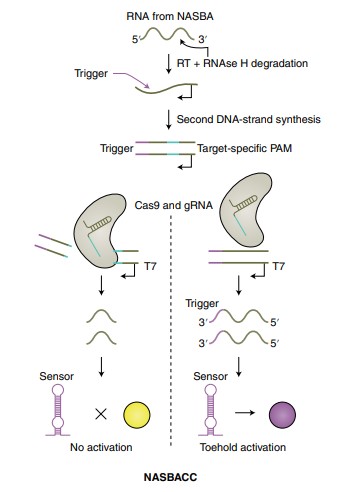
Otro método basado en CRISPR-Cas9 que ha tenido bastante éxito es NASBACC (siglas en inglés para nucleic sequence-based amplification (NASBA)-CRISPR cleavage)36. Este diseño es bastante complejo y combina una técnica de amplificación de fragmentos como es NASBA (que se basa en primers para la amplificación isotérmica continua de ácidos nucleicos), con la propiedad de Cas9 de producir cortes en secuencias diana, de modo que se pueda escindir selectivamente DNA objetivo. El proceso comienza con la amplificación mediante NASBA de las secuencias RNA que deseemos testar. En este paso de amplificación, se produce la transcripción inversa (RT por sus siglas en inglés) a DNA complementario gracias a un primer que tiene ligado consigo una secuencia activadora o trigger. El RNA del producto híbrido DNA/RNA fruto de dicha amplificación es destruido por RNasa H, lo cual hace que a esa cadena simple de DNA que nos queda se le pueda unir un cebador que contiene un promotor T7 y se cree una segunda cadena DNA complementaria. La transcripción de T7 origina una secuencia RNA, la cual puede ser usada como punto de partida para un nuevo ciclo de NASBA. Además, estas secuencias RNA transcritas pueden ser detectadas por un sensor toehold, también denominado toehold switch sensor, unos riboreguladores programados sintéticamente capaces de unirse a las secuencias trigger del RNA transcrito y cuya activación puede provocar un cambio de color en la reacción fácilmente reconocible a simple vista. Sin embargo, aquí entra en juego Cas9, puesto que si hay secuencias PAM en los amplicones dsDNA, Cas9 procederá al corte de los mismos, con lo que se genera un sustrato de dsDNA mucho más pequeño para la transcripción de T7, de modo que la transcripción de estos RNA no activarán el sensor toehold. En definitiva: en ausencia de PAM, sí se produce la activación de los sensores toehold al ser completas las secuencias de RNA fruto de la transcripción, pero si hay secuencias PAM, Cas9 impedirá dicha activación. Un resultado positivo, entendido este como la producción de un cambio en la coloración, indicará que no está presente la secuencia que se está buscando, mientras que un resultado negativo determina que sí está presente tal secuencia. Este método fue testado para detectar virus Zika (ZIKV), obteniendo unos excelentes resultados de especificidad al punto que se consiguió la distinción entre cepas americanas y africanas del virus, con sensibilidad en rango femtomolar36.
Otro diseño más reciente basado en CRISPR-Cas9 es LEOPARD (por leveraging engineered tracrRNAs and on-target DNAs for parallel RNA detection)37, que puede proporcionar la detección multiplex de diferentes secuencias RNA. Conseguir este tipo de detección con estos sistemas es una característica, como luego veremos, que se busca con mucho interés en los últimos años. LEOPARD se basa en reprogramar tracrRNAs para unirse a transcripciones celulares de interés. Esto se debe a la capacidad que tienen algunos tracrRNAs de unirse a las secuencias de RNA (que, como se explicó anteriormente, era algo indispensable para formar la maquinaria procariota de interferencia CRISPR-Cas9). El diseño aprovecha esta característica para hacer que se hibriden tracRNAs a crRNAs no canónicos (ncrRNAs) de interés; su unión podrá conformar entonces un híbrido tracRNA:ncrRNA que dirija a ortólogos de Cas9 a objetivos de DNA complementarios que alberguen PAM, de modo similar a un crRNA canónico. Ninguno de los ncrRNA tendría un PAM en situación correcta, por lo que no se esperaría que los ncrRNA dirijan a Cas9 para escindir su sitio genómico de origen. De este modo, gracias a los ncrRNA y la actividad nucleasa de Cas9 podremos realizar cortes en secuencias de interés, pudiendo detectar esa actividad.
En el caso de todos estos diseños basados en Cas9, esta proteína es usada generalmente para adherirse a la secuencia objetivo, o para realizar cortes en dicha secuencia, pero no posee otras particularidades que la haga más protagonista en el proceso de detección11. De hecho, es importante destacar que la mayor parte de los diseños diagnósticos basados en CRISPR-Cas no han aportado ventajas significativas con respecto a otros métodos de diagnóstico molecular como la PCR. Es por ello que muchos han quedado relegados a ciertas áreas de investigación2. Sin embargo, existen algunos métodos que sí han supuesto una verdadera revolución y un salto adelante; son los métodos basados en la detección mediante las proteínas Cas12 y Cas13, las cuales poseen unas particularidades que las hacen únicas e ideales para usar en estos sistemas diagnósticos.
Destacar antes de entrar a hablar de los diagnósticos basados en CRISPR-Cas12 y Cas13 que existe un único diseño diagnóstico que emplee un sistema CRISPR-Cas de la clase 1. Recordemos que estos sistemas añaden un grado de complejidad por el mayor número de proteínas que emplean, por lo que es más complicado usarlos tanto para la ingeniería genética como para técnicas diagnósticas. Este ensayo se denomina CONAN (siglas de Cas3-Operated Nucleic Acid detection)38 emplea Cas3, que exhibe cierta actividad colateral cuando se une a determinadas secuencias.
| Cas12a | Cas13 | |
| Requerimiento de PAM | Sí | No |
| Secuencia PAM | TTTV | No aplicable |
| Tipo de diana | ssDNA, dsDNA | ssRNA |
| Actividad colateral | Sí | Sí |
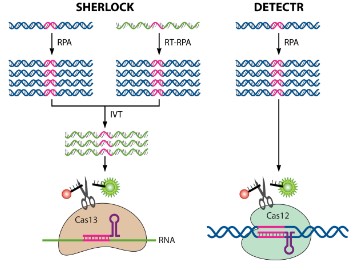
El ensayo DETECTR (siglas para DNA endonuclease-targeted CRISPR trans reporter)28 fue diseñado en 2018 por J. Doudna y colaboradores. Uno de los pasos más importantes en su desarrollo fue el descubrimiento por parte de este equipo de investigación de que un ortólogo de Cas12, LbCas12a (obtenida de Lachnospiraceae bacterium) posee actividad colateral no específica. Esto viene a significar que si se produce el reconocimiento y la unión a la molécula DNA diana, LbCas12a procederá a degradar de manera no específica cualquier molécula adyacente (lo que se ha denominado trans-cleavage), además de realizar cortes en la secuencia diana (lo que se ha denominado cis-cleavage). El diseño aprovecha esta importante característica añadiendo a esta reacción sondas de DNA diseñadas de manera similar a las sondas TaqMan, que en un extremo tienen unido un fluoróforo y en el otro extremo un desactivador de fluorescencia (quencher). De esta manera, cuando LbCas12a procede con su actividad colateral cortará estas sondas, se romperá la proximidad entre fluoróforo y quencher y se producirá una fuerte señal de fluorescencia que podrá ser detectada mediante diversos métodos28.
La sensibilidad aportada por esta reacción alcanza un rango picomolar, lo cual podría tener ciertas limitaciones en algunos requerimientos diagnósticos, como es el caso del control de la carga viral del VIH durante el tratamiento antirretroviral11. La solución a estos problemas, como se describirá más adelante, podría ser el empleo previo a esta reacción de una técnica de amplificación, bien la técnica de PCR, bien técnicas isotérmicas. DETECTR se empleó en el momento de su desarrolló para la detección del Virus Papiloma Humano (HPV), así como para diferenciar entre HPV16 y HPV18, que son los tipos de HPV más pro-oncogénicos. En extractos crudos de DNA, DETECTR identificó HPV16 en 25 de 25 casos y HPV18 en 23 de 25 (los casos habían sido identificados anteriormente mediante PCR)28.
Se han empleado varios ortólogos de Cas12 en otros diseños, tales como AsCas12a (Acidaminococcus sp.) o FnCas12a (Francisella novicida)40, presentando también estas actividad colateral. Es importante destacar que el mismo grupo de trabajo caracterizó la proteína Cas1427, la cual pertenece al tipo V y presenta también actividad colateral. Esta proteína es pequeña, mucho más que otras proteínas Cas y no depende de PAM para poder ejercer su función. Una ventaja de esta proteína es que posee una menor tolerancia a las discordancias de nucleótidos, lo que es fundamental para detectar SNPs (siglas de single nucleotide polymorphisms, que en español se traduce como polimorfismos puntuales o de un solo nucleótido), factor que sería de gran ayuda a la hora de detectar ciertas especies bacterianas multirresistentes2,40. Un diseño basado en DETECTR pero empleando esta enzima consiguió resultados altamente específicos comparados con otros diseños en la detección del gen HERC2 humano, lo que hace a esta enzima una excelente candidata para ser usada para genotipado27.
Existen otras técnicas basadas en proteínas Cas12 que siguen un diseño DETECTR pero que albergan algunas particularidades. HOLMES (siglas para one-hour low-cost multipurpose highly efficient system)41 conlleva un paso previo de amplificación con PCR y emplea LbCas12a unida a sgRNA para detectar DNA/RNA viral, consiguiendo una sensibilidad en rango atomolar en un proceso que conlleva 1 hora. Una versión posterior, HOLMESv242, desecha el paso previo de amplificación con PCR y emplea una amplificación isotérmica con LAMP (siglas de loop-mediated isothermal amplification), además de AscCas12b (obtenida de Alicyclobacillus acidoterrestris), una proteína Cas12b que es termoestable, lo que hace posible combinar los 2 pasos en 1, simplificando el proceso. Cas12b además demostró que podía actuar de manera independiente de PAM, exhibiendo actividad colateral cuando se producía el reconocimiento de ciertas secuencias RNA diana. Se ha demostrado, además, que Cas12b presenta una actividad enzimática mayor en secuencias de dsDNA que Cas12a42.
Usando HOLMES y HOLMESv2 se consiguieron niveles de sensibilidad en rango atomolar para la determinación de virus como el de la encefalitis japonesa, con un Límite de Detección (LOD) en torno a 10 aM. HOLMESv2 contaba además con la ventaja de recortar tiempo al emplear LAMP, consiguiendo resultados en apenas 30 minutos43.
El otro gran avance en el campo del diagnóstico con estas técnicas se dio en 2017 con la creación por parte de F. Zhang y colaboradores de SHERLOCK (siglas para specific high-sensitivity enzymatic reporter unlocking)39. En este caso la proteína Cas que se usa es Cas13, perteneciente al tipo VI de la clase 2 de los sistemas CRISPR-Cas. El esquema general es muy parecido a DETECTR: la secuencia diana es reconocida por un complejo formado por Cas13 y sgRNA y, una vez se ha producido el reconocimiento y la unión, Cas13 despliega su actividad colateral y es capaz de degradar moléculas adyacentes, en este caso de RNA (puesto que las únicas dianas de Cas13 son secuencias ssRNA). SHERLOCK emplea LwaCas13a, proteína Cas obtenida de Leptotrichia wadeii. Lassondas con el fluoróforo y el quencher en este caso deben ser, por tanto, sondas de ssRNA. Además, SHERLOCK también incorpora un paso previo de amplificación, el cual se realiza mediante una técnica isotérmica como RPA (por Recombinase polymerase amplification) con el fin de aumentar la sensibilidad. En caso de que la secuencia que se desee examinar con este diseño sea DNA, en la amplificación mediante RPA se debe usar un primer que añada un promotor T7 al amplicon. Este promotor permite la transcripción a RNA, lo que es necesario para que Cas13 pueda ejercer su actividad39. SHERLOCK fue testado, en el momento de su diseño, para detectar varios virus (Zika y Dengue) y algunas bacterias patógenas, con un LOD en torno a 10-18 M39.
A la hora de hacer comparativas entre los diseños basados en Cas12 y Cas13, debemos resaltar que los ensayos basados en Cas12 producen resultados más rápidos gracias a que sus dianas son moléculas de DNA, lo que va en detrimento de Cas13. Sin embargo, la actividad colateral de Cas12 es menor con respecto a Cas13, por lo que estos diseños aportan mejores datos de sensibilidad y especificidad11,39. Además, no dependen de PAM para ejercer su función, lo que ha convertido a estos diseños como los grandes favoritos cuando se quiere desarrollar una técnica diagnóstica que detecte una secuencia diana que carezca de PAM. Una gran ventaja de SHERLOCK es que varios de sus componentes pueden ser liofilizados y almacenados durante largos periodos de tiempo, lo que posibilita aún más su empleo como POC35,39.
El mismo equipo de trabajo creó posteriormente SHERLOCKv244, un diseño que aportaba la siempre interesante ventaja de la detección multiplexada de ácidos nucleicos. Para poder lograrlo, se emplearon 4 ortólogos de Cas13: LwaCas13a, PsmCas13b, CcaCas13 y AsCas12a, que realizan ejercen cortes mediante su actividad colateral con mayor preferencia en unos u otros dinucleótidos (AU, UC, AC y GA, respectivamente). Este diseño se aprovecha de esta característica para emplear 4 tipos diferentes de sondas, siendo cada uno de ellos ricos en unos u otros dinucleótidos y estando cada tipo de sondas marcado con un fluoróforo distinto (FAM, TEX615, Cy5 y HEX). Esto permite la creación de 4 canales de fluorescencia diferentes, con la detección al mismo tiempo de 4 dianas. Señalar, por último, que SHERLOCKv2 también requeriría de un paso de amplificación previo para aumentar la sensibiilidad, pero se propuso un diseño especialmente innovador que evitaría incluso una amplificación isotérmica. Para ello se combinó LwaCas13a con Csm6, una RNasa que está involucrada en el proceso de interferencia del sistema CRISPR-Cas de tipo III15. Una vez que LwaCas13a corte tanto la secuencia diana como las sondas con fluoróforo, se producirán cOAs, que sirven como segundos mensajeros para activar a Csm6. Esta activación hace que esta proteína realice cortes no específicos en sondas diferentes a las que corta LwaCas13a, ya que son ricas en otros nucleótidos. De este modo se produce señal de fluorescencia por el corte de dos tipos de sondas, incrementándose la sensibilidad unas 3,5 veces con respecto a no emplear Csm6. Este diseño representa un salto enorme en este campo diagnóstico, aportando unas ventajas significativas y es que realmente la necesidad de un paso previo de amplificación para aumentar la sensibilidad de estos ensayos es uno de los mayores retos que se presentan actualmente en este campo2,11.
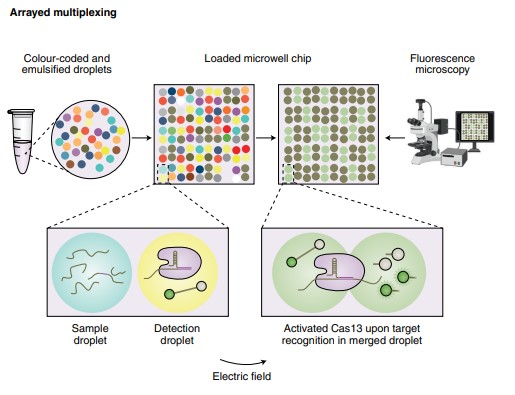
Por otra parte, el diseño multiplex que proporcionan diseños como SHERLOCKv2 ha sido investigado en los últimos años por la gran ventaja que supone el poder detectar varias dianas al mismo tiempo. Fruto de estos esfuerzos es el diseño CARMEN (por combinatorial arrayed reactions for multiplexed evaluation of nucleic acids)45, diseño que permite la identificación multiplexada de 4,500 dianas al mismo tiempo. CARMEN utiliza el diseño SHERLOCK (empleando LwaCas13), pero incorpora tecnología microfluídica, con emulsiones nanolítricas en formas de gotas que, por un lado, contienen las secuencias que se desean analizar, amplificadas previamente mediante PCR o RPA, denominándose a estas emulsiones “gotas de muestra”. Por otro lado, se emplean otras emulsiones que contienen los reactivos de detección; diferentes maquinarias CRISPR-Cas necesarias para distintas detecciones (es decir, diferentes sgRNAs) y diferentes sondas; son las llamadas “gotas de detección”. Las gotas se combinan entre sí y con soluciones de aceite fluorado de distintos colores para que cada una tenga asignada un código visualmente reconocible. A continuación, esas gotas son dispuestas en un microwell-array chip al que se aplicará una corriente eléctrica, de modo que las gotas se fusionan, permitiendo así la reacción CRISPR-Cas basada en SHERLOCK. Si hay actividad colateral, se liberará fluorescencia, siendo detectable por un microscopio de fluorescencia. Esta técnica presenta enormes y obvias ventajas, permitiendo una detección rápida y de bajo coste, con un gran número de posibles combinaciones. Con esta técnica se consiguió la detección simultánea de 169 virus en el momento de su diseño. Aunque conlleve la necesidad de una extracción de ácidos nucleicos muy prolongada (en torno a 8 horas)45, el alto rendimiento de la detección puede paliar ese hecho. Además, la tecnología CARMEN abarata los costes propios de SHERLOCK35. Por otra parte, presenta también la desventaja de ser dependiente de la microscopía, lo que implica, en última instancia, que es una técnica dependiente del observador.
Otro campo de investigación abierto en relación a estas técnicas diagnósticas es cómo evitar el riesgo de contaminaciones que pueden producirse en estos diseños, ya que en muchos casos se incluyen diversos pasos de pipeteo2,11,35. La idea más recurrente en investigación es la de conseguir un diseño que permita llevar a cabo todas las reacciones en un mismo tubo de reacción. Es así como surgieron diseños como OR-DETECTR y OR-SHERLOCK46, que se basan en las reacciones de estos diseños pero incluyendo todos los procesos en un solo paso. Esto se consigue incluyendo tanto la reacción de amplificación como las proteínas Cas en un mismo tubo de reacción. En el extremo superior del tubo de reacción se encontraría el material genético extraído de la muestra y los componentes necesarios para la amplificación isotérmica. En el extremo inferior se encuentran los componentes necesarios (proteínas Cas, sgRNA y sondas con fluoróforos) para la detección de los ácidos nucleicos. Una vez producida la amplificación, se realizaría un simple paso de centrifugación y se mezclarían todos los componentes, para que se produjese así la siguiente parte del proceso.
Una pregunta más a resolver fue la de conseguir la posibilidad de realizar determinaciones sin necesidad de un proceso de extracción, sino que se pudiera realizar la detección de ácidos nucleicos directamente de la muestra (como saliva, orina u otros líquidos biológicos)11. El método HUDSON (por heating unextracted diagnostic samples to obliterate nucleases)47 intenta dar respuesta a esta pregunta. Este método utiliza un proceso de calor y reducción química que se aplica directamente a la muestra para inactivar los altos niveles de nucleasas presentes en fluidos corporales y para conseguir la lisis de partículas virales, liberando así ácidos nucleicos, pudiendo detectarlos con SHERLOCK. Todo este proceso conlleva poco tiempo y fue testado experimentalmente, consiguiéndose detecciones de los virus Zika (ZIKV) y virus Dengue en muestras de sangre, suero y saliva en un tiempo de 2 horas, con excelentes resultados de sensibilidad (se consiguió detecciones a concentraciones tan bajas como 1 copia/µl)48. También se ha empleado HUDSON para la detección de virus de fiebres hemorrágicas49. Por último, empleando HUDSON y SHERLOCK ha nacido SHINE (por streamiline highlighting of infectious to navigate pandemics)50, diseñado a raíz de la pandemia por SARS-CoV2 para la detección de este virus en muestras de exudados nasofaríngeos y en salvia, usando HUDSON para acelerar la extracción de viral en las muestras. Este diseño consiguió cifras de sensibilidad del 90% y de especificidad del 100% con respecto a la PCR50.
En línea con el diseño POC que se intenta incorporar a estas pruebas diagnósticas, la mayor parte de todos estos diseños buscan métodos de lectura de resultados simples, baratos y fáciles de interpretar11. Existen diversos métodos de lectura y entre ellos es relevante destacar los métodos basados en la detección de la liberación de la fluorescencia tras la ruptura de las sondas que tienen ligados fluoróforos, cuya lectura se puede realizar con la simple visualización a simple vista bajo luz azul, con microscopios de fluorescencia o con lectores de fluorescencia. Por otro lado, uno de los sistemas más utilizados2,11,35, por las evidentes ventajas que aporta, es el lateral flow device (LFD) basado en inmunocromatografía. Un LFD que se emplea en la lectura de un diseño SHERLOCKv244 se basa en la alta afinidad que presenta la estreptavidina por la biotina, de modo que usa una sonda de DNA o RNA que tiene ligado un fluoróforo y biotina. El producto de la reacción entre el material genético extraído de la muestra y la maquinaria de reacción Cas13/sgRNA se deposita en la zona de aplicación de la muestra de la tira, donde están presentes unas nanopartículas de oro, que tienen ligadas anticuerpo anti-FITC (isotiocinato de fluoresceína). FAM se unirá a estos anticuerpos, de modo que por medio de una acción capilar todo el complejo viajará a lo largo de la tira del LFD. Existe una primera banda donde está presente estreptavidina, que se unirá a la biotina. Si no se ha producido reacción entre el material extraído de la muestra y la maquinaria Cas13/sgRNA (esto es, si no están presentes en la muestras las secuencias que se deseen detectar), la sonda con fluoróforo y biotina estará intacta al no haberse cortado por la actividad colateral de Cas. Esto hace que todo el complejo, FAM incluido, queden anclados en esta primera banda, indicando un resultado negativo. En cambio, si se ha producido actividad colateral de Cas al estar presente la secuencia que se deseaba detectar, la sonda estará rota, de modo que mientras que la biotina queda anclada a la estreptavidina en la primera banda, el complejo conformado por FAM, los anticuerpos anti-FITC a los que está unidos y las nanopartículas de oro que están a su vez unidas a los anticuerpos seguirán discurriendo por el LFD, hasta alcanzar una segunda banda más lejana de anticuerpos. Estos anticuerpos se unen a los anticuerpos anti-FITC, quedando aquí anclado el complejo, resultando en una segunda banda que indica un resultado positivo.
Otro método de lectura de resultados interesante a destacar es un método eléctrico basado en hidrogeles de DNA51. El hidrogel consiste en un polímero que contiene moléculas de DNA que sirven como ancla y elemento estructural del gel. En caso de que exista reconocimiento de las secuencias a detectar por Cas12/sgRNA, la actividad colateral de Cas12 cortará las moléculas de DNA del gel, modificando las propiedades del mismo. De esta manera se produce una degradación del gel, impidiendo la reticulación del mismo, que sí se produce cuando no hay actividad colateral de Cas12 (esto es, cuando la prueba es negativa). Al impedirse la reticulación del gel, se permitirá el flujo de un buffer a través del mismo, flujo que puede ser leído mediante un dispositivo de tecnología microfluídica basada en papel multicapa (µPAD)51,52. Este sistema va a permitir, por tanto, que podamos hacer tanto una análisis visual del resultado (observando a simple vista si está presente o no el flujo del buffer) como una lectura eléctrica, ya que el flujo pasará a través de los canales del µPAD y se puede hacer una medición gracias a la disposición de dos electrodos, que miden la cantidad de flujo, indicando la cantidad de secuencia diana presente en la reacción. En resumen: a mayor actividad colateral de Cas12, mayor rotura de DNA en el hidrogel, por lo que mayor flujo y mayor intensidad de señal de eléctrica.
En cuanto a la colorimetría también existen métodos de lectura basados en nanopartículas de oro unidas a sondas dsDNA:ssDNA53. Estas sondas tienen unidas 2 nanopartículas; cuando se produce la actividad colateral por Cas12 al detectar secuencia diana, se corta la cadena ssDNA de la sonda, lo que aleja ambas nanopartículas de oro. La separación entre ambas nanopartículas modifica el color de la solución de rojo a rojo púrpura. Este ensayo ha sido probado en un campo distinto al de las enfermedades infecciosas; en la detección de genes responsables del cancer hereditario de mama y ovario (BRCA1), pero los autores sugieren otros usos también.

Por último hay que destacar que algunos diseños se combinan con técnicas de NGS (next-generation sequencing)11. Uno de los más interesantes es FLASH (finding low abundance sequences by hibridization)54, que emplea Cas9 para realizar cortes en secuencias de interés y es un diseño bastante interesante en caso de disponer de escasas cantidades de la muestra que se desee secuenciar. FLASH comienza con un primer paso de bloqueo mediante fosfatasa de las terminaciones de todas las secuencias DNA de una muestra. A continuación CRISPR-Cas9 realizará cortes en las secuencias de interés, cortes que originan fragmentos cuyos extremos no están bloqueados, por lo que se les puede unir adaptadores que permitan la realización de una PCR, la cual amplificará únicamente las secuencias de interés, aumentando su concentración en 5 órdenes de magnitud. Estos fragmentos se podrán usar a continuación para la secuenciación de nueva generación por medio de Illumina. En el momento de su diseño los autores usaron la técnica para la detección y secuenciación, en fluidos respiratorios y en muestras de sangre, de bacterias Gram positivas multirresistentes en concentraciones subatomolares y para la detección de Plasmodium falciparum54. Los autores proponen el uso de esta técnica para la detección altamente multiplexada de genes de resistencia en muestras clínicas, aunque también proponen su uso futuro para estudio de microbioma y otros54.
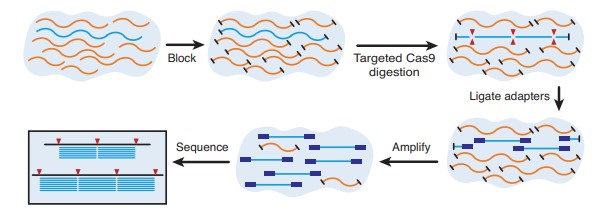
Otro diseño que emplea CRISPR-Cas combinado con NGS es DASH (por depletion of abundant sequences by hybridation)55, que sigue una esquemática parecida a FLASH pero en sentido opuesto: usando Cas9 se consigue la eliminación de secuencias que no sean un objetivo para su detección antes de proceder con la técnica de secuenciación, consiguiendo de esa manera aumentar la concentración de secuencias de patógenos, desechando las secuencias que no interesen. DASH requiere la construcción de una librería de crRNAs que dirijan a las proteínas Cas9 hacia esas secuencias no deseadas. Finalmente, existen otros diseños como nCATS (por nanopore Cas9-targeted sequencing)56 o STRique (por short tandem repeat identification, quantification and evaluation)57 que combinan un método de enriquecimiento de secuencias mediante Cas9 con la secuenciación a través de nanoporos.
APLICACIONES DE LOS SISTEMAS DIAGNÓSTICOS BASADOS EN CRISPR-CAS
Estos sistemas de diagnóstico han ido creciendo en número y complejidad en un corto espacio de tiempo2,11. El campo principal de aplicación diagnóstica de estos sistemas han sido las enfermedades infecciosas, pero no el único, dado que se han usado para el diagnóstico de diversos tipos de cáncer o para el de algunas enfermedades hereditarias35,58. Como se ha comentado anteriormente, muchos de estos ensayos tienen como objetivo el empleo de técnicas diagnósticas baratas, sencillas y de fácil uso, más idóneas para países con bajos recursos que tengan mayores dificultades a la hora de reunir un equipo adecuado. Además, estas regiones pueden estar más sujetas a problemas de almacenaje de determinados componentes o problemas para la adquisición de determinados reactivos. Todos estos inconvenientes pueden imposibilitar o dificultar el control de brotes epidémicos; por tanto es fundamental el desarrollo de nuevas técnicas diagnósticas que aporten una solución o, por lo menos, contribuyan a la lucha contra las epidemias. Las técnicas diagnósticas basadas en CRISPR-Cas pueden ser una respuesta a esos problemas.
El área de mayor aplicación de estas técnicas dentro del campo de las enfermedades infecciosas ha sido la detección de patógenos virales. Como se mencionó anteriormente, en su nacimiento el diseño DETECTR fue aplicado para la detección y diferenciación de HPV28. Existen otros diseños creados con ese mismo fin de detección de HPV, como es el diseño CARP59, ctPCR3.060 o CDetection61, siendo este uno de los patógenos en los que más se han centrado los diseños de esta tecnología. CARP (CRISPR-associated reverse PCR) y ctPCR (CRISPR-typing PCR) usan PCR como método de amplificación. En concreto ctPCR emplea 2 complejos CRISPR-Cas9 para dirigirse hacia una secuencia de elección, para así poder añadir cebadores a los extremos de esa secuencia una vez cortada y proceder con la PCR.
Otro patógeno para cuya detección se han creado numerosos ensayos es SARS-CoV2, coincidiendo con la reciente pandemia mundial. De hecho, para la detección de este patógeno disponemos del único ensayo aprobado por la FDA en mayo de 202062, un diseño basado en SHERLOCK que incorporaba un paso previo de amplificación mediante LAMP. Este ensayo posee un LOD de 42 copias de RNA por reacción y ofrecía la posibilidad de lecturas mediante fluorescencia y lateral-flow, siendo la sensibilidad mayor con la primera opción que con la segunda62. La detección se realizaba de muestras nasofaríngeas y exudados faríngeos. La especificidad que se consiguió fue del 100% y el tiempo necesario para obtener el resultado fue en torno a 70 minutos62. Otros diseños para SARS-CoV2 son el ya mencionado SHINE50 o STOPCovid63, que implica un diseño con un paso previo de extracción de RNA y amplificación isotérmica mediante LAMP, además de usar una proteína Cas termoestable (AapCas12b), ya que todo el proceso se realiza en un solo tubo de reacción. STOPCovid ofreció buenos resultados de sensibilidad en ensayos con muestras nasofaríngeas, comparables a la RT-qPCR, con un LOD de 100 copias de genoma viral por reacción y una sensibilidad del 95%, resultando el proceso de reacción en 1 hora63. SHINE por otra parte obtuvo buenos resultados en ensayos con muestras nasofaríngeas en una cifra de tiempo muy destacable: 50 minutos, con un 90% de sensibilidad y un 100% de especificidad comparado con qPCR50. También es reseñable en relación a SARS-CoV2 el diseño AIOD-CRISPR (por all-in-one dual CRISPR-Cas12a)64, que usa una maquinaria compuesta por Cas12a siguiendo un diseño DETECTR. Las enzimas empleadas en este diseño, curiosamente, no son dependientes de PAM y este diseño, que también innova con la ventaja de que todas las reacciones sucedan en el mismo tubo de ensayo, alcanza un LOD de 5 copias/reacción. En un ensayo este diseño fue capaz de encontrar todas las muestras positivas (8, detectadas previamente por qPCR) de un total de 28 exudados nasofaríngeos64.
Otros virus también incluidos en diseños diagnósticos han sido el virus del Ebola, Dengue y Zika, algunos ya comentados, estando la mayor parte de ellos basados en NASBACC, SHERLOCK y variantes de este mismo esquema que empleaban HUDSON49. En el caso de los virus de las fiebres hemorrágicas se hace imprescindible que el diseño de la técnica diagnóstica sea sencillo y barato, dada el área geográfica donde suelen producirse estos brotes; zonas donde la implementación de un diagnóstico con PCR puede ser complicado. En este aspecto, SHERLOCKv2 se ha demostrado como el mejor de estos sistemas para la detección de Dengue49.
También existen diseños CRISPR-Cas, basados en SHERLOCK, para la detección de virus de la familia Herpesviridae (como el citomegalovirus (CMV)65) o de la familia Poliomaviridae (BK virus)65. En el caso de este último virus, se testaron 36 muestras de plasma y 31 muestras de orinas, arrojando el experimento cifras del 100% de sensibilidad y especificidad65.
En cuanto a las bacterias detectadas por estas técnicas, la más utilizada ha sido Mycobacterium tuberculosis. Así por ejemplo disponemos de un ensayo, mencionado anteriormente, basado en Cas9 (dCas9)33 o un diseño basado en Cas12a66. Con este último ensayo, se estudiaron muestras de 179 pacientes con tuberculosis, y se obtuvieron datos de sensibilidad del 79%, más alto que con el cultivo o con otros métodos de diagnóstico molecular66. Existe otro ensayo denominado TB-QUICK67 diseñado muy recientemente para acortar el tiempo de resultado en torno a 2 horas, con un LOD de 1,3 copias/µl y con una sensibilidad para muestras pulmonares del 86,8%, también más alto que el cultivo y otras técnicas.
Otras bacterias detectadas han sido Listeria monocytogenes (diseño denominado CASLFA, por CRISPR-Cas9 mediated lateral flow nucleic acid assay68, basado en Cas9 y usando LFD como método de lectura, proporcionando resultados en 1 hora), Salmonella enteritidis67 o Staphylococcus aureus44,48,69, con resultados variables en cuanto a sensibilidad y especificidad. Concretamente F. Zhang y su grupo de investigación han desarrollado un diseño SHERLOCKv2 para la detección multiplex de S. aureus y P. aeruginosa que alcanza un LOD de rango atomolar44.
En cuanto a protozoos se han diseñado ensayos para la detección de protozoos del grupo Plasmodium, de entre los que podemos destacar uno basado en Cas12a, con amplificación previa mediante RPA70. También se puede resaltar en este aspecto un diseño de SHERLOCK que es capaz de detectar todas las especies de Plasmodium causantes de malaria71. Se realizaron pruebas con 123 muestras clínicas y se obtuvo un resultado de un 94% de sensibilidad y especificidad (comparado con qPCR)71. Es importante recordar que FLASH también se empleó en el momento de su diseño para la detección de este protozoo54.
Otra aplicación importante de estos sistemas diagnósticos es la detección de polimorfismos de nucleótidos únicos (SNPs, por sus siglas en inglés)2,11,58. La detección de estas mutaciones en una secuencia nos lleva a la posibilidad de usar esta tecnología CRISPR-Cas para la detección de marcadores de la resistencia antimicrobiana70, siendo muchos de estos marcadores mutaciones en secuencias claves sobre las que un antibiótico ejerce su función o que codifican proteínas indispensables para la actuación de un antibiótico.
CONCLUSIONES
La tecnología CRISPR-Cas ha constituido una de las mayores revoluciones en el campo de la biología y la biotecnología de las últimas décadas3. Su aplicación en el campo del diagnóstico microbiológico es incluso más reciente que otras aplicaciones, pero se han llegado a auténticos avances en muy poco tiempo, especialmente en lo que a su diseño como POC se refiere35. Si además comparamos con la tecnología PCR, los diseños diagnósticos basados CRISPR-Cas son mucho más baratos, simples, transportables y rápidos; cuanto más recientes son los diseños más se pueden observar estas diferencias2,11.
Pero quedan obstáculos por franquear: se requiere mayor experimentación en la práctica clínica habitual que permita hacer comparaciones con otros sistemas de diagnóstico molecular como la PCR2,11,35,38,58. Además es necesario este uso en la práctica real que permita establecer como de aplicables son estos diseños, así como la calidad de diferentes muestras biológicas.
Por otra parte existe el riesgo de falsos positivos que puedan surgir de un reconocimiento erróneo de las secuencias dianas, dado que algunos sistemas CRISPR-Cas son capaces de tolerar ciertas alteraciones de nucleótidos, surgiendo reconocimientos off-targets, que, aunque infrecuentes, son posibles35.
Otro gran obstáculo, en el que en este escrito se ha insistido varias veces, es la necesidad de un paso previo de amplificación para alcanzar buenos rangos de sensibilidad2,11,35, lo que aleja a estas técnicas de la configuración de POC ideal. Incluso aún en un diseño como OR-SHERLOCK, sigue siendo necesario el completar 2 pasos diferenciados, lo que puede hacer que algunos de los componentes no trabajen en sus condiciones ideales35. Por otra parte, las técnicas de amplificación isotérmicas, si bien más sencillas, conllevan una sensibilidad y una especificidad menor que la PCR. La vía a mejorar en estos casos sería una mayor optimización de las proteínas Cas que se usen, así como una mejora en las técnicas de amplificación.
En cuanto a la sencillez y rapidez de los ensayos, son 2 de sus mayores ventajas11,35. Algunos tiempos de reacción son solo de 15 minutos, mientras que las técnicas suelen ser bastante sencillas para los operarios11 (aunque puede haber excepciones como el diseño CARMEN). El método de lectura también puede ser muy sencillo, siendo portátil en muchos de los diseños y habiendo cada vez más ensayos que optan por simplificar ese proceso. El coste, por otra parte, si bien no está aún definido11, dado que muchos test no están comercializados, se supone más bajo que con respecto a otras técnicas, aunque sigue siendo una incógnita. La posibilidad de liofilización de SHERLOCK y otros ensayos es otro de los grandes atractivos11,35.
También es necesario un mayor desarrollo del campo de la bioinformática en relación al diseño de sgRNAs, con el fin de obtener una amplia librería de dianas, lo que puede conllevar un aumento de la especificidad de estos diseños35.
Todas estas desventajas deben ser puestas en una balanza con las ventajas para obtener una panorámica de la situación actual y el futuro de estos ensayos. Aún queda mucho campo de estudio: la automatización de la tecnología CRISPR para el diagnóstico es un requisito casi indispensable, como se ha podido comprobar durante la pandemia por SARS-CoV2 y el gran volumen de muestras que se han recibido en los laboratorios de Microbiología. Integrar los conceptos de POC con una automatización en un solo paso parece a la vez una oportunidad y un reto; conseguir un sistema que fuese capaz de realizar las diversas etapas del proceso diagnóstico en un solo aparato con la máxima fiabilidad parece ser hacia donde se dirigen los esfuerzos de los nuevos diseños.
Es improbable que estas técnicas desbanquen a la PCR en un plazo corto o medio de tiempo2, pero pueden hacerse un lugar en el abanico de técnicas diagnósticas que un laboratorio de Microbiología Clínica puede ofrecer. Además, se ha abierto un nuevo campo de investigación lleno de posibilidades para el desarrollo de herramientas con las que luchar contra las enfermedades infecciosas.
BIBLIOGRAFÍA
- Peeling RW, Holmes KK, Mabey D, Ronald A. Rapid tests for sexually transmitted infections (STIs): the way forward. Sex Transm Infect. 2006 Dec;82 Suppl 5(Suppl 5):v1-6. doi: 10.1136/sti.2006.024265. Epub 2006 Dec 6. PMID: 17151023; PMCID: PMC2563912.
- Kostyusheva A, Brezgin S, Babin Y, Vasilyeva I, Glebe D, Kostyushev D, Chulanov V. CRISPR-Cas systems for diagnosing infectious diseases. Methods. 2022 Jul;203:431-446. doi: 10.1016/j.ymeth.2021.04.007. Epub 2021 Apr 9. PMID: 33839288; PMCID: PMC8032595.
- Lander ES. The Heroes of CRISPR. Cell. 2016 Jan 14;164(1-2):18-28. doi: 10.1016/j.cell.2015.12.041. PMID: 26771483.
- Baker RE, Mahmud AS, Miller IF, Rajeev M, Rasambainarivo F, Rice BL, Takahashi S, Tatem AJ, Wagner CE, Wang LF, Wesolowski A, Metcalf CJE. Infectious disease in an era of global change. Nat Rev Microbiol. 2022 Apr;20(4):193-205. doi: 10.1038/s41579-021-00639-z. Epub 2021 Oct 13. PMID: 34646006; PMCID: PMC8513385.
- Dip SD, Sarkar SL, Setu MAA, Das PK, Pramanik MHA, Alam ASMRU, Al-Emran HM, Hossain MA, Jahid IK. Evaluation of RT-PCR assays for detection of SARS-CoV-2 variants of concern. Sci Rep. 2023 Feb 9;13(1):2342. doi: 10.1038/s41598-023-28275-y. PMID: 36759632; PMCID: PMC9910272.
- Mahony JB, Blackhouse G, Babwah J, Smieja M, Buracond S, Chong S, Ciccotelli W, O’Shea T, Alnakhli D, Griffiths-Turner M, Goeree R. Cost analysis of multiplex PCR testing for diagnosing respiratory virus infections. J Clin Microbiol. 2009 Sep;47(9):2812-7. doi: 10.1128/JCM.00556-09. Epub 2009 Jul 1. PMID: 19571025; PMCID: PMC2738055.
- Woodford N, Ellington MJ. The emergence of antibiotic resistance by mutation. Clin Microbiol Infect. 2007 Jan;13(1):5-18. doi: 10.1111/j.1469-0691.2006.01492.x. PMID: 17184282.
- Doudna JA, Charpentier E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science. 2014 Nov 28;346(6213):1258096. doi: 10.1126/science.1258096. PMID: 25430774.
- Wang H, La Russa M, Qi LS. CRISPR/Cas9 in Genome Editing and Beyond. Annu Rev Biochem. 2016 Jun 2;85:227-64. doi: 10.1146/annurev-biochem-060815-014607. Epub 2016 Apr 25. PMID: 27145843.
- Yang H, Wang H, Jaenisch R. Generating genetically modified mice using CRISPR/Cas-mediated genome engineering. Nat Protoc. 2014 Aug;9(8):1956-68. doi: 10.1038/nprot.2014.134. Epub 2014 Jul 24. PMID: 25058643.
- Kaminski MM, Abudayyeh OO, Gootenberg JS, Zhang F, Collins JJ. CRISPR-based diagnostics. Nat Biomed Eng. 2021 Jul;5(7):643-656. doi: 10.1038/s41551-021-00760-7. Epub 2021 Jul 16. PMID: 34272525.
- Mustafa MI, Makhawi AM. SHERLOCK and DETECTR: CRISPR-Cas Systems as Potential Rapid Diagnostic Tools for Emerging Infectious Diseases. J Clin Microbiol. 2021 Feb 18;59(3):e00745-20. doi: 10.1128/JCM.00745-20. PMID: 33148705; PMCID: PMC8106734.
- Rostøl JT, Marraffini L. (Ph)ighting Phages: How Bacteria Resist Their Parasites. Cell Host Microbe. 2019 Feb 13;25(2):184-194. doi: 10.1016/j.chom.2019.01.009. PMID: 30763533; PMCID: PMC6383810.
- Bernheim A, Sorek R. The pan-immune system of bacteria: antiviral defence as a community resource. Nat Rev Microbiol. 2020 Feb;18(2):113-119. doi: 10.1038/s41579-019-0278-2. Epub 2019 Nov 6. PMID: 31695182.
- Hille F, Richter H, Wong SP, Bratovič M, Ressel S, Charpentier E. The Biology of CRISPR-Cas: Backward and Forward. Cell. 2018 Mar 8;172(6):1239-1259. doi: 10.1016/j.cell.2017.11.032. PMID: 29522745.
- Goldfarb T, Sberro H, Weinstock E, Cohen O, Doron S, Charpak-Amikam Y, Afik S, Ofir G, Sorek R. BREX is a novel phage resistance system widespread in microbial genomes. EMBO J. 2015 Jan 13;34(2):169-83. doi: 10.15252/embj.201489455. Epub 2014 Dec 1. PMID: 25452498; PMCID: PMC4337064.
- Ofir G, Melamed S, Sberro H, Mukamel Z, Silverman S, Yaakov G, Doron S, Sorek R. DISARM is a widespread bacterial defence system with broad anti-phage activities. Nat Microbiol. 2018 Jan;3(1):90-98. doi: 10.1038/s41564-017-0051-0. Epub 2017 Oct 30. PMID: 29085076; PMCID: PMC5739279.
- Makarova KS, Wolf YI, Alkhnbashi OS, Costa F, Shah SA, Saunders SJ, Barrangou R, Brouns SJ, Charpentier E, Haft DH, Horvath P, Moineau S, Mojica FJ, Terns RM, Terns MP, White MF, Yakunin AF, Garrett RA, van der Oost J, Backofen R, Koonin EV. An updated evolutionary classification of CRISPR-Cas systems. Nat Rev Microbiol. 2015 Nov;13(11):722-36. doi: 10.1038/nrmicro3569. Epub 2015 Sep 28. PMID: 26411297; PMCID: PMC5426118.
- Hille F, Charpentier E. CRISPR-Cas: biology, mechanisms and relevance. Philos Trans R Soc Lond B Biol Sci. 2016 Nov 5;371(1707):20150496. doi: 10.1098/rstb.2015.0496. PMID: 27672148; PMCID: PMC5052741.
- Brouns SJ, Jore MM, Lundgren M, Westra ER, Slijkhuis RJ, Snijders AP, Dickman MJ, Makarova KS, Koonin EV, van der Oost J. Small CRISPR RNAs guide antiviral defense in prokaryotes. Science. 2008 Aug 15;321(5891):960-4. doi: 10.1126/science.1159689. PMID: 18703739; PMCID: PMC5898235.
- Hayes RP, Xiao Y, Ding F, van Erp PB, Rajashankar K, Bailey S, Wiedenheft B, Ke A. Structural basis for promiscuous PAM recognition in type I-E Cascade from E. coli. Nature. 2016 Feb 25;530(7591):499-503. doi: 10.1038/nature16995. Epub 2016 Feb 10. PMID: 26863189; PMCID: PMC5134256.
- Kazlauskiene M, Kostiuk G, Venclovas Č, Tamulaitis G, Siksnys V. A cyclic oligonucleotide signaling pathway in type III CRISPR-Cas systems. Science. 2017 Aug 11;357(6351):605-609. doi: 10.1126/science.aao0100. Epub 2017 Jun 29. PMID: 28663439.
- Jinek M, Jiang F, Taylor DW, Sternberg SH, Kaya E, Ma E, Anders C, Hauer M, Zhou K, Lin S, Kaplan M, Iavarone AT, Charpentier E, Nogales E, Doudna JA. Structures of Cas9 endonucleases reveal RNA-mediated conformational activation. Science. 2014 Mar 14;343(6176):1247997. doi: 10.1126/science.1247997. Epub 2014 Feb 6. PMID: 24505130; PMCID: PMC4184034.
- Jiang F, Doudna JA. CRISPR-Cas9 Structures and Mechanisms. Annu Rev Biophys. 2017 May 22;46:505-529. doi: 10.1146/annurev-biophys-062215-010822. Epub 2017 Mar 30. PMID: 28375731.
- Yang H, Gao P, Rajashankar KR, Patel DJ. PAM-Dependent Target DNA Recognition and Cleavage by C2c1 CRISPR-Cas Endonuclease. Cell. 2016 Dec 15;167(7):1814-1828.e12. doi: 10.1016/j.cell.2016.11.053. PMID: 27984729; PMCID: PMC5278635.
- Yamano T, Nishimasu H, Zetsche B, Hirano H, Slaymaker IM, Li Y, Fedorova I, Nakane T, Makarova KS, Koonin EV, Ishitani R, Zhang F, Nureki O. Crystal Structure of Cpf1 in Complex with Guide RNA and Target DNA. Cell. 2016 May 5;165(4):949-62. doi: 10.1016/j.cell.2016.04.003. Epub 2016 Apr 21. PMID: 27114038; PMCID: PMC4899970.
- Harrington LB, Burstein D, Chen JS, Paez-Espino D, Ma E, Witte IP, Cofsky JC, Kyrpides NC, Banfield JF, Doudna JA. Programmed DNA destruction by miniature CRISPR-Cas14 enzymes. Science. 2018 Nov 16;362(6416):839-842. doi: 10.1126/science.aav4294. Epub 2018 Oct 18. PMID: 30337455; PMCID: PMC6659742.
- Chen JS, Ma E, Harrington LB, Da Costa M, Tian X, Palefsky JM, Doudna JA. CRISPR-Cas12a target binding unleashes indiscriminate single-stranded DNase activity. Science. 2018 Apr 27;360(6387):436-439. doi: 10.1126/science.aar6245. Epub 2018 Feb 15. Erratum in: Science. 2021 Feb 19;371(6531): PMID: 29449511; PMCID: PMC6628903.
- East-Seletsky A, O’Connell MR, Knight SC, Burstein D, Cate JH, Tjian R, Doudna JA. Two distinct RNase activities of CRISPR-C2c2 enable guide-RNA processing and RNA detection. Nature. 2016 Oct 13;538(7624):270-273. doi: 10.1038/nature19802. Epub 2016 Sep 26. PMID: 27669025; PMCID: PMC5576363.
- Sashital DG, Jinek M, Doudna JA. An RNA-induced conformational change required for CRISPR RNA cleavage by the endoribonuclease Cse3. Nat Struct Mol Biol. 2011 Jun;18(6):680-7. doi: 10.1038/nsmb.2043. Epub 2011 May 15. PMID: 21572442.
- Zhang Y, Heidrich N, Ampattu BJ, Gunderson CW, Seifert HS, Schoen C, Vogel J, Sontheimer EJ. Processing-independent CRISPR RNAs limit natural transformation in Neisseria meningitidis. Mol Cell. 2013 May 23;50(4):488-503. doi: 10.1016/j.molcel.2013.05.001. PMID: 23706818; PMCID: PMC3694421.
- Koonin EV, Makarova KS, Zhang F. Diversity, classification and evolution of CRISPR-Cas systems. Curr Opin Microbiol. 2017 Jun;37:67-78. doi: 10.1016/j.mib.2017.05.008. Epub 2017 Jun 9. PMID: 28605718; PMCID: PMC5776717.
- Zhang Y, Wang Y, Xu L, Lou C, Ouyang Q, Qian L. Paired dCas9 design as a nucleic acid detection platform for pathogenic strains. Methods. 2022 Jul;203:70-77. doi: 10.1016/j.ymeth.2021.06.003. Epub 2021 Jun 4. PMID: 34090973.
- Strich JR, Chertow DS. CRISPR-Cas Biology and Its Application to Infectious Diseases. J Clin Microbiol. 2019 Mar 28;57(4):e01307-18. doi: 10.1128/JCM.01307-18. PMID: 30429256; PMCID: PMC6440769.
- Li L, Shen G, Wu M, Jiang J, Xia Q, Lin P. CRISPR-Cas-mediated diagnostics. Trends Biotechnol. 2022 Nov;40(11):1326-1345. doi: 10.1016/j.tibtech.2022.04.006. Epub 2022 May 17. PMID: 35595574.
- Pardee K, Green AA, Takahashi MK, Braff D, Lambert G, Lee JW, Ferrante T, Ma D, Donghia N, Fan M, Daringer NM, Bosch I, Dudley DM, O’Connor DH, Gehrke L, Collins JJ. Rapid, Low-Cost Detection of Zika Virus Using Programmable Biomolecular Components. Cell. 2016 May 19;165(5):1255-1266. doi: 10.1016/j.cell.2016.04.059. Epub 2016 May 6. PMID: 27160350.
- Jiao C, Sharma S, Dugar G, Peeck NL, Bischler T, Wimmer F, Yu Y, Barquist L, Schoen C, Kurzai O, Sharma CM, Beisel CL. Noncanonical crRNAs derived from host transcripts enable multiplexable RNA detection by Cas9. Science. 2021 May 28;372(6545):941-948. doi: 10.1126/science.abe7106. Epub 2021 Apr 27. PMID: 33906967; PMCID: PMC8224270.
- Yoshimi K, Takeshita K, Yamayoshi S, Shibumura S, Yamauchi Y, Yamamoto M, Yotsuyanagi H, Kawaoka Y, Mashimo T. CRISPR-Cas3-based diagnostics for SARS-CoV-2 and influenza virus. iScience. 2022 Feb 18;25(2):103830. doi: 10.1016/j.isci.2022.103830. Epub 2022 Jan 30. PMID: 35128347; PMCID: PMC8801231.
- Kellner MJ, Koob JG, Gootenberg JS, Abudayyeh OO, Zhang F. SHERLOCK: nucleic acid detection with CRISPR nucleases. Nat Protoc. 2019 Oct;14(10):2986-3012. doi: 10.1038/s41596-019-0210-2. Epub 2019 Sep 23. Erratum in: Nat Protoc. 2020 Mar;15(3):1311. PMID: 31548639; PMCID: PMC6956564.
- Makarova KS, Wolf YI, Iranzo J, Shmakov SA, Alkhnbashi OS, Brouns SJJ, Charpentier E, Cheng D, Haft DH, Horvath P, Moineau S, Mojica FJM, Scott D, Shah SA, Siksnys V, Terns MP, Venclovas Č, White MF, Yakunin AF, Yan W, Zhang F, Garrett RA, Backofen R, van der Oost J, Barrangou R, Koonin EV. Evolutionary classification of CRISPR-Cas systems: a burst of class 2 and derived variants. Nat Rev Microbiol. 2020 Feb;18(2):67-83. doi: 10.1038/s41579-019-0299-x. Epub 2019 Dec 19. PMID: 31857715; PMCID: PMC8905525.
- Li SY, Cheng QX, Wang JM, Li XY, Zhang ZL, Gao S, Cao RB, Zhao GP, Wang J. CRISPR-Cas12a-assisted nucleic acid detection. Cell Discov. 2018 Apr 24;4:20. doi: 10.1038/s41421-018-0028-z. Erratum in: Cell Discov. 2019 Mar 12;5:17. PMID: 29707234; PMCID: PMC5913299.
- Li L, Li S, Wu N, Wu J, Wang G, Zhao G, Wang J. HOLMESv2: A CRISPR-Cas12b-Assisted Platform for Nucleic Acid Detection and DNA Methylation Quantitation. ACS Synth Biol. 2019 Oct 18;8(10):2228-2237. doi: 10.1021/acssynbio.9b00209. Epub 2019 Sep 30. PMID: 31532637.
- Wang B, Wang R, Wang D, Wu J, Li J, Wang J, Liu H, Wang Y. Cas12aVDet: A CRISPR/Cas12a-Based Platform for Rapid and Visual Nucleic Acid Detection. Anal Chem. 2019 Oct 1;91(19):12156-12161. doi: 10.1021/acs.analchem.9b01526. Epub 2019 Sep 11. PMID: 31460749.
- Gootenberg JS, Abudayyeh OO, Kellner MJ, Joung J, Collins JJ, Zhang F. Multiplexed and portable nucleic acid detection platform with Cas13, Cas12a, and Csm6. Science. 2018 Apr 27;360(6387):439-444. doi: 10.1126/science.aaq0179. Epub 2018 Feb 15. PMID: 29449508; PMCID: PMC5961727.
- Ackerman CM, Myhrvold C, Thakku SG, Freije CA, Metsky HC, Yang DK, Ye SH, Boehm CK, Kosoko-Thoroddsen TF, Kehe J, Nguyen TG, Carter A, Kulesa A, Barnes JR, Dugan VG, Hung DT, Blainey PC, Sabeti PC. Massively multiplexed nucleic acid detection with Cas13. Nature. 2020 Jun;582(7811):277-282. doi: 10.1038/s41586-020-2279-8. Epub 2020 Apr 29. PMID: 32349121; PMCID: PMC7332423.
- Sun Y, Yu L, Liu C, Ye S, Chen W, Li D, Huang W. One-tube SARS-CoV-2 detection platform based on RT-RPA and CRISPR/Cas12a. J Transl Med. 2021 Feb 16;19(1):74. doi: 10.1186/s12967-021-02741-5. PMID: 33593370; PMCID: PMC7884969.
- Myhrvold C, Freije CA, Gootenberg JS, Abudayyeh OO, Metsky HC, Durbin AF, Kellner MJ, Tan AL, Paul LM, Parham LA, Garcia KF, Barnes KG, Chak B, Mondini A, Nogueira ML, Isern S, Michael SF, Lorenzana I, Yozwiak NL, MacInnis BL, Bosch I, Gehrke L, Zhang F, Sabeti PC. Field-deployable viral diagnostics using CRISPR-Cas13. Science. 2018 Apr 27;360(6387):444-448. doi: 10.1126/science.aas8836. PMID: 29700266; PMCID: PMC6197056.
- Gootenberg JS, Abudayyeh OO, Lee JW, Essletzbichler P, Dy AJ, Joung J, Verdine V, Donghia N, Daringer NM, Freije CA, Myhrvold C, Bhattacharyya RP, Livny J, Regev A, Koonin EV, Hung DT, Sabeti PC, Collins JJ, Zhang F. Nucleic acid detection with CRISPR-Cas13a/C2c2. Science. 2017 Apr 28;356(6336):438-442. doi: 10.1126/science.aam9321. Epub 2017 Apr 13. PMID: 28408723; PMCID: PMC5526198.
- Barnes KG, Lachenauer AE, Nitido A, Siddiqui S, Gross R, Beitzel B, Siddle KJ, Freije CA, Dighero-Kemp B, Mehta SB, Carter A, Uwanibe J, Ajogbasile F, Olumade T, Odia I, Sandi JD, Momoh M, Metsky HC, Boehm CK, Lin AE, Kemball M, Park DJ, Branco L, Boisen M, Sullivan B, Amare MF, Tiamiyu AB, Parker ZF, Iroezindu M, Grant DS, Modjarrad K, Myhrvold C, Garry RF, Palacios G, Hensley LE, Schaffner SF, Happi CT, Colubri A, Sabeti PC. Deployable CRISPR-Cas13a diagnostic tools to detect and report Ebola and Lassa virus cases in real-time. Nat Commun. 2020 Aug 17;11(1):4131. doi: 10.1038/s41467-020-17994-9. PMID: 32807807; PMCID: PMC7431545.
- Arizti-Sanz J, Freije CA, Stanton AC, Petros BA, Boehm CK, Siddiqui S, Shaw BM, Adams G, Kosoko-Thoroddsen TF, Kemball ME, Uwanibe JN, Ajogbasile FV, Eromon PE, Gross R, Wronka L, Caviness K, Hensley LE, Bergman NH, MacInnis BL, Happi CT, Lemieux JE, Sabeti PC, Myhrvold C. Streamlined inactivation, amplification, and Cas13-based detection of SARS-CoV-2. Nat Commun. 2020 Nov 20;11(1):5921. doi: 10.1038/s41467-020-19097-x. PMID: 33219225; PMCID: PMC7680145.
- English MA, Soenksen LR, Gayet RV, de Puig H, Angenent-Mari NM, Mao AS, Nguyen PQ, Collins JJ. Programmable CRISPR-responsive smart materials. Science. 2019 Aug 23;365(6455):780-785. doi: 10.1126/science.aaw5122. PMID: 31439791.
- English MA, Soenksen LR, Gayet RV, de Puig H, Angenent-Mari NM, Mao AS, Nguyen PQ, Collins JJ. Programmable CRISPR-responsive smart materials. Science. 2019 Aug 23;365(6455):780-785. doi: 10.1126/science.aaw5122. PMID: 31439791.
- Yin X, Yang H, Piao Y, Zhu Y, Zheng Q, Khan MR, Zhang Y, Busquets R, Hu B, Deng R, Cao J. CRISPR-Based Colorimetric Nucleic Acid Tests for Visual Readout of DNA Barcode for Food Authenticity. J Agric Food Chem. 2022 Nov 2;70(43):14052-14060. doi: 10.1021/acs.jafc.2c05974. Epub 2022 Oct 24. PMID: 36278890.
- Quan J, Langelier C, Kuchta A, Batson J, Teyssier N, Lyden A, Caldera S, McGeever A, Dimitrov B, King R, Wilheim J, Murphy M, Ares LP, Travisano KA, Sit R, Amato R, Mumbengegwi DR, Smith JL, Bennett A, Gosling R, Mourani PM, Calfee CS, Neff NF, Chow ED, Kim PS, Greenhouse B, DeRisi JL, Crawford ED. FLASH: a next-generation CRISPR diagnostic for multiplexed detection of antimicrobial resistance sequences. Nucleic Acids Res. 2019 Aug 22;47(14):e83. doi: 10.1093/nar/gkz418. PMID: 31114866; PMCID: PMC6698650.
- Gu W, Crawford ED, O’Donovan BD, Wilson MR, Chow ED, Retallack H, DeRisi JL. Depletion of Abundant Sequences by Hybridization (DASH): using Cas9 to remove unwanted high-abundance species in sequencing libraries and molecular counting applications. Genome Biol. 2016 Mar 4;17:41. doi: 10.1186/s13059-016-0904-5. PMID: 26944702; PMCID: PMC4778327.
- Gilpatrick T, Lee I, Graham JE, Raimondeau E, Bowen R, Heron A, Downs B, Sukumar S, Sedlazeck FJ, Timp W. Targeted nanopore sequencing with Cas9-guided adapter ligation. Nat Biotechnol. 2020 Apr;38(4):433-438. doi: 10.1038/s41587-020-0407-5. Epub 2020 Feb 10. PMID: 32042167; PMCID: PMC7145730.
- Giesselmann P, Brändl B, Raimondeau E, Bowen R, Rohrandt C, Tandon R, Kretzmer H, Assum G, Galonska C, Siebert R, Ammerpohl O, Heron A, Schneider SA, Ladewig J, Koch P, Schuldt BM, Graham JE, Meissner A, Müller FJ. Analysis of short tandem repeat expansions and their methylation state with nanopore sequencing. Nat Biotechnol. 2019 Dec;37(12):1478-1481. doi: 10.1038/s41587-019-0293-x. Epub 2019 Nov 18. PMID: 31740840.
- Binnie A, Fernandes E, Almeida-Lousada H, de Mello RA, Castelo-Branco P. CRISPR-based strategies in infectious disease diagnosis and therapy. Infection. 2021 Jun;49(3):377-385. doi: 10.1007/s15010-020-01554-w. Epub 2021 Jan 3. PMID: 33393066; PMCID: PMC7779109.
- Zhang B, Wang Q, Xu X, Xia Q, Long F, Li W, Shui Y, Xia X, Wang J. Detection of target DNA with a novel Cas9/sgRNAs-associated reverse PCR (CARP) technique. Anal Bioanal Chem. 2018 May;410(12):2889-2900. doi: 10.1007/s00216-018-0873-5. Epub 2018 Mar 15. PMID: 29546544.
- Zhang B, Xia Q, Wang Q, Xia X, Wang J. Detecting and typing target DNA with a novel CRISPR-typing PCR (ctPCR) technique. Anal Biochem. 2018 Nov 15;561-562:37-46. doi: 10.1016/j.ab.2018.09.012. Epub 2018 Sep 19. PMID: 30243976.
- Teng F, Guo L, Cui T, Wang XG, Xu K, Gao Q, Zhou Q, Li W. CDetection: CRISPR-Cas12b-based DNA detection with sub-attomolar sensitivity and single-base specificity. Genome Biol. 2019 Jul 1;20(1):132. doi: 10.1186/s13059-019-1742-z. PMID: 31262344; PMCID: PMC6604390.
- Zhu X, Wang X, Li S, Luo W, Zhang X, Wang C, Chen Q, Yu S, Tai J, Wang Y. Rapid, Ultrasensitive, and Highly Specific Diagnosis of COVID-19 by CRISPR-Based Detection. ACS Sens. 2021 Mar 26;6(3):881-888. doi: 10.1021/acssensors.0c01984. Epub 2021 Mar 1. PMID: 33645226; PMCID: PMC7945583.
- Joung J, Ladha A, Saito M, Kim NG, Woolley AE, Segel M, Barretto RPJ, Ranu A, Macrae RK, Faure G, Ioannidi EI, Krajeski RN, Bruneau R, Huang MW, Yu XG, Li JZ, Walker BD, Hung DT, Greninger AL, Jerome KR, Gootenberg JS, Abudayyeh OO, Zhang F. Detection of SARS-CoV-2 with SHERLOCK One-Pot Testing. N Engl J Med. 2020 Oct 8;383(15):1492-1494. doi: 10.1056/NEJMc2026172. Epub 2020 Sep 16. PMID: 32937062; PMCID: PMC7510942.
- Ding X, Yin K, Li Z, Liu C. All-in-One Dual CRISPR-Cas12a (AIOD-CRISPR) Assay: A Case for Rapid, Ultrasensitive and Visual Detection of Novel Coronavirus SARS-CoV-2 and HIV virus. bioRxiv [Preprint]. 2020 Mar 21:2020.03.19.998724. doi: 10.1101/2020.03.19.998724. Update in: Nat Commun. 2020 Sep 18;11(1):4711. PMID: 32511323; PMCID: PMC7239053.
- Kaminski MM, Alcantar MA, Lape IT, Greensmith R, Huske AC, Valeri JA, Marty FM, Klämbt V, Azzi J, Akalin E, Riella LV, Collins JJ. A CRISPR-based assay for the detection of opportunistic infections post-transplantation and for the monitoring of transplant rejection. Nat Biomed Eng. 2020 Jun;4(6):601-609. doi: 10.1038/s41551-020-0546-5. Epub 2020 Apr 13. PMID: 32284553.
- Ai JW, Zhou X, Xu T, Yang M, Chen Y, He GQ, Pan N, Cai Y, Li Y, Wang X, Su H, Wang T, Zeng W, Zhang WH. CRISPR-based rapid and ultra-sensitive diagnostic test for Mycobacterium tuberculosis. Emerg Microbes Infect. 2019;8(1):1361-1369. doi: 10.1080/22221751.2019.1664939. PMID: 31522608; PMCID: PMC6758691.
- Shen J, Zhou X, Shan Y, Yue H, Huang R, Hu J, Xing D. Sensitive detection of a bacterial pathogen using allosteric probe-initiated catalysis and CRISPR-Cas13a amplification reaction. Nat Commun. 2020 Jan 14;11(1):267. doi: 10.1038/s41467-019-14135-9. PMID: 31937772; PMCID: PMC6959245.
- Wang X, Xiong E, Tian T, Cheng M, Lin W, Wang H, Zhang G, Sun J, Zhou X. Clustered Regularly Interspaced Short Palindromic Repeats/Cas9-Mediated Lateral Flow Nucleic Acid Assay. ACS Nano. 2020 Feb 25;14(2):2497-2508. doi: 10.1021/acsnano.0c00022. Epub 2020 Feb 14. PMID: 32045522.
- Guk K, Keem JO, Hwang SG, Kim H, Kang T, Lim EK, Jung J. A facile, rapid and sensitive detection of MRSA using a CRISPR-mediated DNA FISH method, antibody-like dCas9/sgRNA complex. Biosens Bioelectron. 2017 Sep 15;95:67-71. doi: 10.1016/j.bios.2017.04.016. Epub 2017 Apr 13. PMID: 28412663.
- Cunningham CH, Hennelly CM, Lin JT, Ubalee R, Boyce RM, Mulogo EM, Hathaway N, Thwai KL, Phanzu F, Kalonji A, Mwandagalirwa K, Tshefu A, Juliano JJ, Parr JB. A novel CRISPR-based malaria diagnostic capable of Plasmodium detection, species differentiation, and drug-resistance genotyping. EBioMedicine. 2021 Jun;68:103415. doi: 10.1016/j.ebiom.2021.103415. Epub 2021 Jun 14. PMID: 34139428; PMCID: PMC8213918.
- Lee RA, Puig H, Nguyen PQ, Angenent-Mari NM, Donghia NM, McGee JP, Dvorin JD, Klapperich CM, Pollock NR, Collins JJ. Ultrasensitive CRISPR-based diagnostic for field-applicable detection of Plasmodium species in symptomatic and asymptomatic malaria. Proc Natl Acad Sci U S A. 2020 Oct 13;117(41):25722-25731. doi: 10.1073/pnas.2010196117. Epub 2020 Sep 21. PMID: 32958655; PMCID: PMC7568265.
